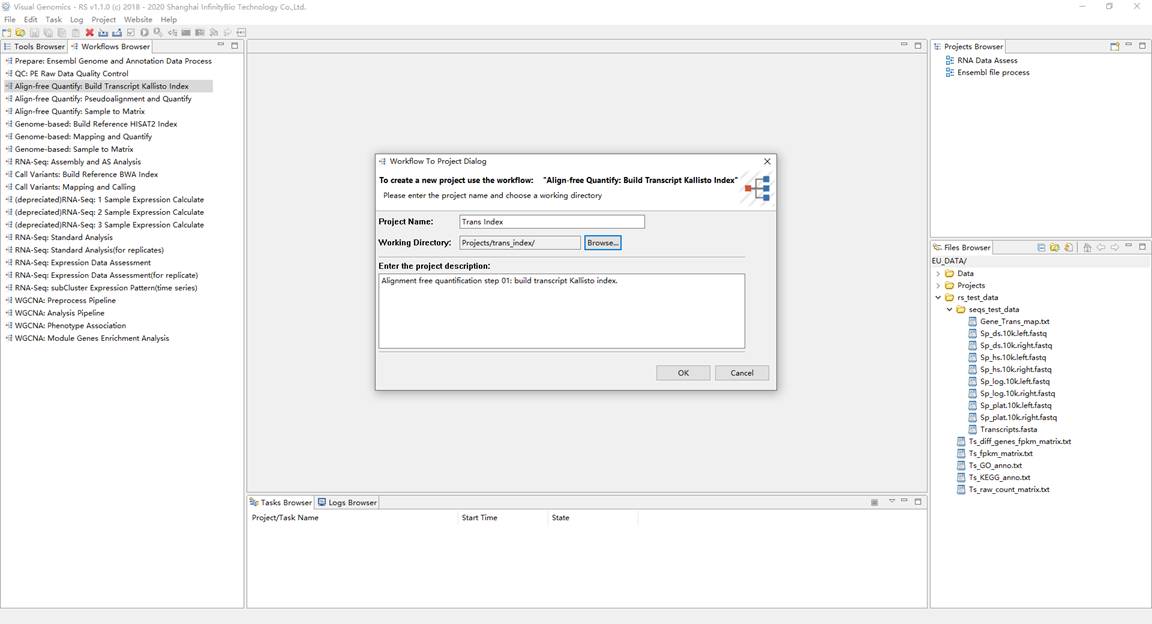
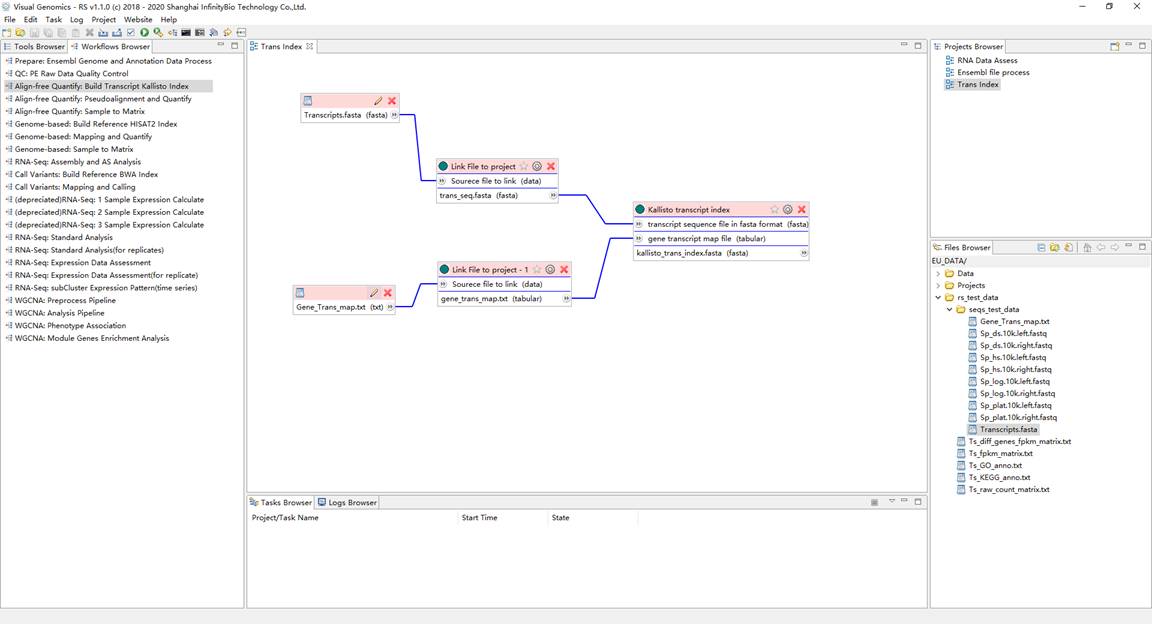
1、建立参考转录本序列索引文件。在Workflows Browser中,双击流程 "Align-free Quantify: Build Transcript Kallisto Index" ,弹出通过流程创建项目对话框,并设置项目名称和工作目录。双击打开项目,将转录本序列 "Transcripts.fasta" 和 "Gene_Trans_map.txt" 作为项目流程的输入,并运行项目得到结果。
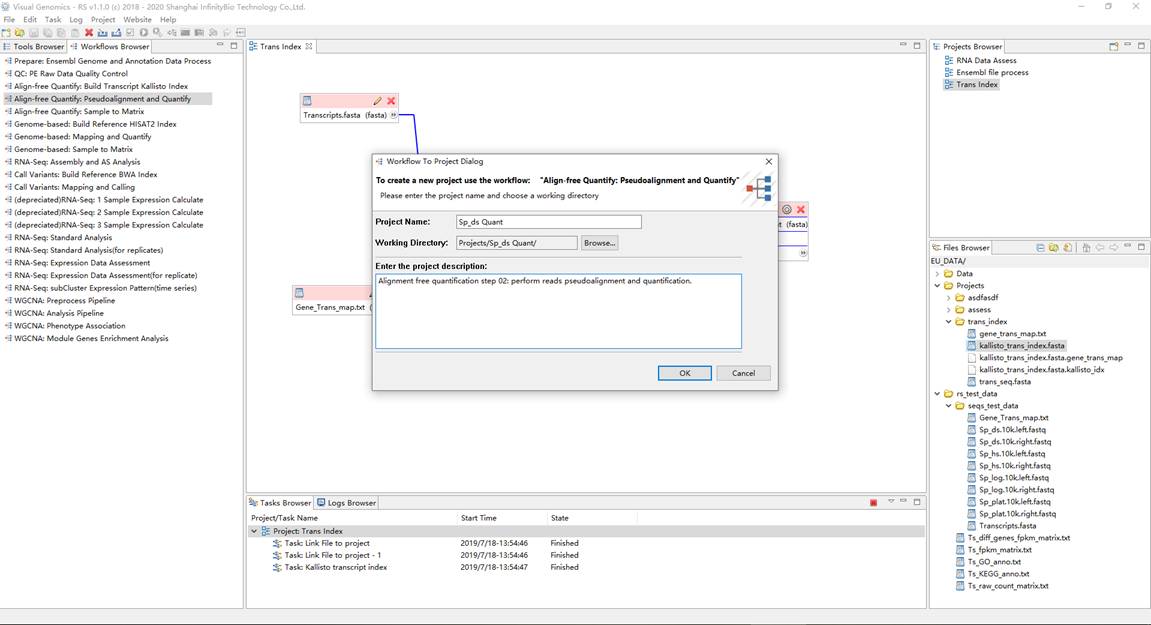
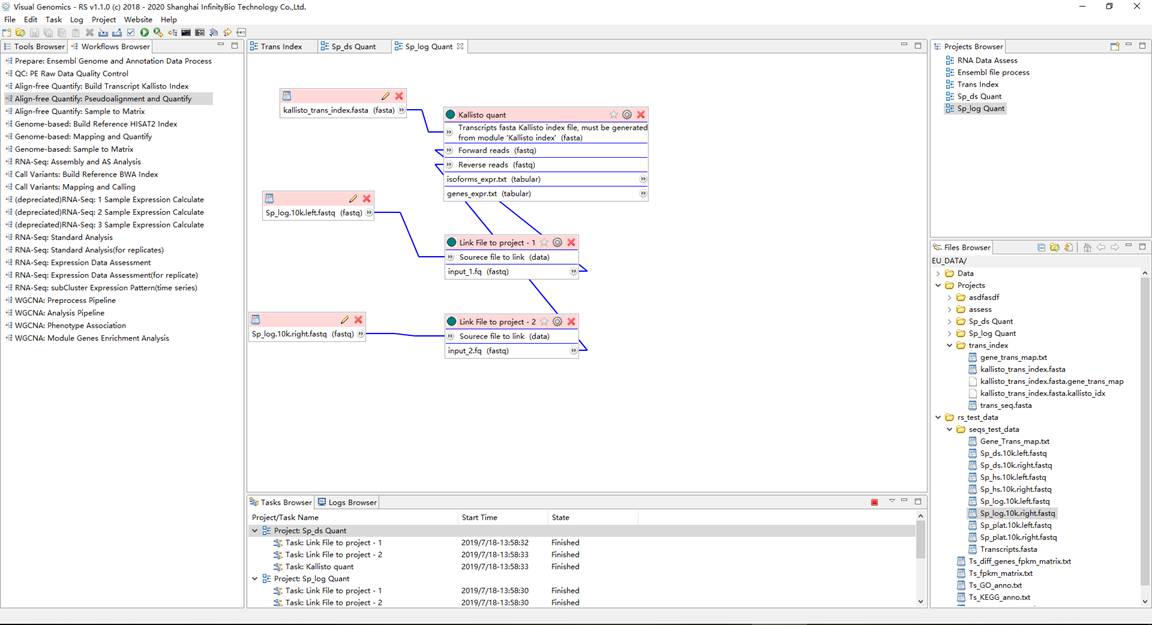
2、定量分析(需要每个样品重复执行该步骤)。在Workflows Browser中,双击流程 "Align-free Quantify: Pseudoalignment and Quantify" ,弹出通过流程创建项目对话框,并设置项目名称和工作目录。双击打开项目, 将上一步得到的转录本索引文件 "kallisto_trans_index.fasta" 和 原始fastq下机数据 作为项目流程的输入, 并运行项目得到结果。这里,每个样品重复运行该步骤,从而得到每个样品的定量结果。需要注意的是,每次新建项 目,均要设置不同的工作目录,避免同名文件覆盖冲突问题。
3、每个样品的定量结果进行合并,从而得到Count矩阵、FPKM矩阵、TPM矩阵。在Workflows Browser中,双击流程 "Align-free Quantify: Sample to Matrix" ,弹出通过流程创建项目对话框,并设置项目名称和工作目录。双击打开 项目,将每个样品的基因定量结果作为项目流程的输入,并运行项目得到结果。
